Syndrome de Pitt-Hopkins
| OMIM | 610954 |
|---|---|
| MeSH | et C537403 C537403 et C537403 |
![]() Mise en garde médicale
Mise en garde médicale
Le syndrome de Pitt-Hopkins (PTHS) est une affection génétique rare se manifestant par un retard de développement et une déficience intellectuelle de modérée à sévère[1], des traits faciaux distinctifs et une possible hyperventilation intermittente suivie d'apnée[2]. L'épilepsie (crises récurrentes) survient souvent dans le cadre du syndrome de Pitt-Hopkins[1]. Elle fait partie du spectre clinique des syndromes de type Rett[3]. Le syndrome de Pitt-Hopkins est cliniquement similaire au syndrome d'Angelman, au syndrome de Rett, au syndrome de Mowat Wilson et au syndrome ATR-X[4].
Au fur et à mesure que les connaissances sur le syndrome de Pitt-Hopkins progressent, le spectre développemental du trouble s'élargit, englobant désormais des difficultés liées à l'anxiété et à l'autisme[5], au TDAH et aux troubles sensoriels. Elle est associée à une anomalie du chromosome 18 qui provoque une expression insuffisante du gène TCF4[6]. Les personnes atteintes du PTHS présentent des taux élevés d’automutilation et de comportements agressifs généralement liés à l’autisme et à leurs troubles sensoriels[7].
Le PTHS a longtemps été associé à une déficience cognitive sévère, mais l'intelligence à proprement parler est difficile à évaluer en raison des troubles moteurs et de la parole. Grâce à l'utilisation de la communication améliorée et alternative et de thérapies plus avancées, de nombreuses personnes parviennent à accomplir bien plus que ce qui avait été initialement envisagé. Il est désormais évident que le spectre des capacités cognitives dans le cadre du syndrome de Pitt-Hopkins est plus large que ce qui est souvent rapporté dans les publications scientifiques. Bien qu'il n'existe pas de traitement curatif pour le syndrome de Pitt-Hopkins, les symptômes associés peuvent être pris en charge[4]. Les chercheurs ont développé des modèles cellulaires et de rongeurs pour tester des thérapies pour la maladie de Pitt-Hopkins[8].
On estime que le PTHS survient chez 1 personne sur 11 000 à 1 personne sur 41 000[9].
Signes et symptômes
Le PTHS peut être observé dès l'enfance[10].
Les premiers signes visibles chez les nourrissons concernent la partie inférieure du visage et la racine nasale haute[9].
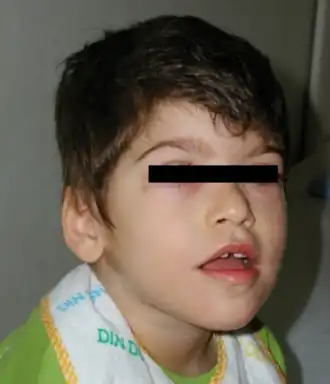
Les traits du visage sont caractéristiques et comprennent[11] :
- Pont nasal large avec pointe bulbeuse
- Large bouche
- Philtrum de l'arc de Cupidon
- Oreilles proéminentes
- Sourcils fins
Des pieds plats, des orteils chevauchants et des coussinets fœtaux sont également courants[11]. Une petite taille et une scoliose sont fréquentes[11].
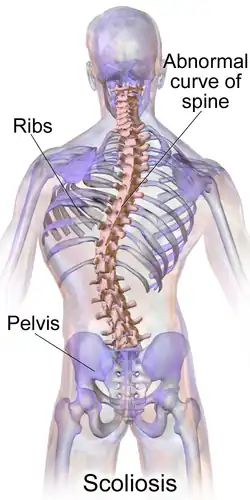
Le syndrome de Pitt-Hopkins peut également se manifester par d'autres caractéristiques telles que la constipation et d'autres troubles gastro-intestinaux, une taille de tête anormalement petite (microcéphalie), de la myopie, un strabisme, une taille réduite et de légères anomalies cérébrales[12].
Les adultes atteints de PTHS peuvent avoir des problèmes d’élocution[10]. Les caractéristiques craniofaciales, qui sont importantes lors du diagnostic du PTHS, deviennent plus visibles à mesure que la personne vieillit[9].
Les enfants atteints du syndrome de Pitt-Hopkins ont généralement un comportement heureux et excitable, avec des sourires fréquents, des rires et des mouvements de battement des mains. Cependant, ils peuvent également souffrir d’anxiété et de problèmes de comportement[12].
Gastro-intestinal
Les troubles gastro-intestinaux sont fréquents chez les personnes atteintes du syndrome de Pitt-Hopkins, incluant la constipation, le reflux et les éructations. La constipation sévère persiste souvent tout au long de la vie. Des problèmes respiratoires peuvent provoquer l'ingestion d'air et des douleurs associées. Un faible tonus musculaire peut également entraîner des difficultés alimentaires dès le plus jeune âge[13].
Neurologique
L’épilepsie est relativement fréquente dans le cadre du syndrome de Pitt-Hopkins, touchant entre 37 et 50 % des cas. Les crises peuvent survenir dès la petite enfance ou à tout âge chez l'adulte. Divers types de crises peuvent être observés, et les résultats des électroencéphalogrammes (EEG) peuvent être soit typiques, soit atypiques, en fonction de chaque individu[13].
L'imagerie par résonance magnétique (IRM) peut montrer des anomalies cérébrales chez les personnes atteintes du syndrome de Pitt-Hopkins, telles qu'un corps calleux de taille réduite, des ventricules dilatés et des anomalies au niveau de la fosse postérieure. Cependant, de nombreuses personnes atteintes de ce syndrome peuvent également présenter des structures cérébrales normales[13].
Musculosquelettique
Des anomalies mineures des mains et des pieds, telles que des membres fins ou petits, des extrémités des doigts larges, une clinodactylie, des doigts effilés, un pli palmaire transversal, des pieds plats avec une déformation en valgus de l'arrière-pied, des orteils chevauchants et des métatarses courts, ont été observées. L'absence de plis de flexion des pouces peut survenir en cas d'ankylose du pouce. Lors d'une intervention chirurgicale, un tendon du pouce absent a également été constaté chez un patient[14].
Génétique
La cause génétique de ce trouble a été décrite en 2007[15]. Ce trouble est dû à une haploinsuffisance du gène du facteur de transcription 4 (TCF4) situé sur le bras long du chromosome 18 (18q21.2). Le spectre mutationnel semble être de 40 % de mutations ponctuelles, 30 % de délétions/insertions mineures et 30 % de délétions. Toutes semblent être des mutations de novo. Le risque d'apparition chez les frères et sœurs est faible, mais plus élevé que dans la population générale en raison du mosaïcisme germinal parental[9].
Un phénotype de type Pitt-Hopkins 1 a été associé à des mutations autosomiques récessives du gène de la protéine associée à la contactine de type 2 (CNTNAP2) situé sur le bras long du chromosome 7 (7q33-q36), tandis qu'un phénotype de type Pitt-Hopkins 2 a été lié à des mutations autosomiques récessives du gène de la neurexine 1 alpha (NRXN1) sur le bras court du chromosome 2 (2p16.3)[16]. Le syndrome de Pitt-Hopkins de type 1 est extrêmement rare, avec une prévalence estimée à moins de 1 sur un million.
Des malformations du système nerveux central peuvent être observées chez environ 60 à 70 % des patients lors d'examens IRM[17].
Les patients atteints du syndrome de Pitt-Hopkins présentant une délétion du gène TCF4 peuvent ne pas présenter les traits faciaux caractéristiques du syndrome[9].
Diagnostic
Il n'existe pas de critères diagnostiques spécifiques, mais certains symptômes peuvent suggérer un diagnostic de PTHS. Parmi ces symptômes figurent la dysmorphie faciale, le retard global du développement avec une apparition précoce, la déficience intellectuelle modérée à sévère, les troubles respiratoires et l'absence d'autres anomalies congénitales majeures[10].
Zollino et ses collègues ont établi des critères diagnostiques fondés sur des caractéristiques observées dans 75 % des cas génétiquement confirmés de PTHS, appelées caractéristiques cardinales. Lorsqu'une personne présente 9 de ces caractéristiques cardinales, elle est diagnostiquée comme étant atteinte de PTHS[13].
Il est possible qu’un phénotype ressemblant au PTHS puisse se produire sans mutation dans le gène TCF4. Les mutations du gène TCF4 n’entraînent pas toujours un syndrome stéréotypé de Pitt-Hopkins[13].
La moitié des personnes atteintes du syndrome de stress post-traumatique souffrent de crises convulsives, dès l'enfance jusqu'à la fin de l'adolescence[9].
Environ 50 % des personnes concernées présentent des anomalies à l’imagerie cérébrale. Il s'agit notamment d'un corps calleux hypoplasique avec un rostre et une partie postérieure du splénium manquants, avec des noyaux caudés bulbeux bombés vers les cornes frontales[4]
Selon le diagnostic clinique. Le PTHS fait partie du même groupe que les troubles envahissants du développement[18].
Lorsqu'un patient est suspecté d'être atteint du PTHS, des tests génétiques examinant le gène TCF4 sont généralement effectués[9]. Certains préconisent un test génétique préalable, suivi d'une évaluation clinique[4].
Diagnostic différentiel
La symptomatologie du PTHS est similaire à celle du syndrome d'Angelman, du syndrome de Rett et du syndrome de Mowat-Wilson[17].
Le syndrome d'Angelman présente des similitudes avec le PTHS, notamment l'absence de langage et un tempérament "joyeux". Parmi les diagnostics différentiels, le syndrome de Rett est le moins similaire au PTHS, étant classé comme une encéphalopathie progressive. Contrairement au PTHS, le syndrome d'Angelman et le syndrome de Rett ne présentent pas les traits faciaux distinctifs. Le syndrome de Mowat-Wilson, quant à lui, se manifeste dès la petite enfance et est caractérisé par des anomalies faciales spécifiques[17].
Traitement
Il n'existe pas de traitement spécifique pour cette affection, la prise en charge étant axée sur les symptômes. En l'absence de traitement curatif, les personnes atteintes du PTHS recourent à des approches comportementales et éducatives[18]. Les comorbidités peuvent également être traitées[4].
Les soins peuvent être assurés par une équipe médicale composée de neurologues, d'ophtalmologues, de pneumologues et de gastro-entérologues[4].
Recommandations pour le retard de développement et la déficience intellectuelle aux États-Unis (peuvent différer selon les pays)[9] :
- Un programme d'intervention précoce, couvrant la période du nouveau-né jusqu'à 3 ans, offrira l'accès à diverses thérapies, telles que l'ergothérapie, la physiothérapie, l'orthophonie et les soins alimentaires.
- De l'école maternelle au système scolaire public, de 3 à 5 ans, l'enfant devra d'abord passer une évaluation afin de déterminer le type de thérapie nécessaire avant d'intégrer le programme.
- Entre 5 et 21 ans, l'école où l'enfant se trouve scolarisé peut élaborer un plan d'éducation individualisé adapté à ses fonctions et à ses besoins. Les enfants sont encouragés à poursuivre leur scolarité au moins jusqu'à l'âge de 21 ans.
Histoire

La maladie a été décrite pour la première fois en 1978 par D. Pitt et I. Hopkins (The Children's Cottages Training Centre, Kew et Royal Children's Hospital, Melbourne, Australie) chez deux patients non apparentés[19].
Références
- 1 2 David A. Sweetser, Ibrahim Elsharkawi, Lael Yonker, Marcie Steeves, Kimberly Parkin et Ronald Thibert, Pitt-Hopkins Syndrome, Seattle (WA), University of Washington, Seattle, (PMID 22934316, lire en ligne)[réf. incomplète]
- ↑ (en) Christiane Zweier, Maarit M. Peippo, Juliane Hoyer et Sergio Sousa, « Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome) », American Journal of Human Genetics, vol. 80, no 5, , p. 994–1001 (ISSN 0002-9297, PMID 17436255, PMCID 1852727, DOI 10.1086/515583, lire en ligne, consulté le )
- ↑ (en) Sandra Whalen, Delphine Héron, Thierry Gaillon et Oana Moldovan, « Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum », Human Mutation, vol. 33, no 1, , p. 64–72 (ISSN 1098-1004, PMID 22045651, DOI 10.1002/humu.21639, S2CID 9559486, lire en ligne, consulté le )
- 1 2 3 4 5 6 (en) Kimberly Goodspeed, Cassandra Newsom, Mary Ann Morris et Craig Powell, « Pitt-Hopkins Syndrome: A Review of Current Literature, Clinical Approach, and 23-Patient Case Series », Journal of Child Neurology, vol. 33, no 3, , p. 233–244 (ISSN 1708-8283, PMID 29318938, PMCID 5922265, DOI 10.1177/0883073817750490, lire en ligne, consulté le )
- ↑ (en-US) « Pitt-Hopkins syndrome may point the way to autism treatments », sur The Transmitter: Neuroscience News and Perspectives, (consulté le )
- ↑ « Pitt-Hopkins »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?), National Center for Biotechnology Information (consulté le )
- ↑ (en) Alice Watkins, Stacey Bissell, Jo Moss et Chris Oliver, « Behavioural and psychological characteristics in Pitt-Hopkins syndrome: a comparison with Angelman and Cornelia de Lange syndromes », Journal of Neurodevelopmental Disorders, vol. 11, no 1, , p. 24 (ISSN 1866-1955, PMID 31586495, PMCID 6778364, DOI 10.1186/s11689-019-9282-0, lire en ligne, consulté le )
- ↑ (en-US) Daniel R. Weinberger, « A drug for autism? Potential treatment for Pitt-Hopkins syndrome offers clues », The Conversation, (lire en ligne, consulté le )
- 1 2 3 4 5 6 7 8 (en) David A. Sweetser, Ibrahim Elsharkawi, Lael Yonker et Marcie Steeves, « Pitt-Hopkins Syndrome », dans GeneReviews, University of Washington, Seattle, (PMID 22934316, lire en ligne)
- 1 2 3 (en) Laura Dean, « Pitt-Hopkins Syndrome », dans Medical Genetics Summaries, National Center for Biotechnology Information (US), (PMID 28520343, lire en ligne)
- 1 2 3 (en) National Cancer Institute, « Pitt-Hopkins Syndrome », Qeios, (ISSN 2632-3834, DOI 10.32388/NB53OY, S2CID 161843893, lire en ligne, consulté le )
- 1 2 Pitt-Hopkins syndrome, Qeios, (DOI 10.32388/l1967e
 )[réf. incomplète]
)[réf. incomplète] - 1 2 3 4 5 (en) Marcella Zollino, Christiane Zweier, Ingrid D. Van Balkom et David A. Sweetser, « Diagnosis and management in Pitt-Hopkins syndrome: First international consensus statement », Clinical Genetics, vol. 95, no 4, , p. 462–478 (ISSN 1399-0004, PMID 30677142, DOI 10.1111/cge.13506, hdl 2066/201958, lire en ligne, consulté le )
- ↑ David A. Sweetser, Ibrahim Elsharkawi, Lael Yonker, Marcie Steeves, Kimberly Parkin et Ronald Thibert, Pitt-Hopkins Syndrome, Seattle (WA), University of Washington, Seattle, (PMID 22934316, lire en ligne)[réf. incomplète]
- ↑ (en) Jeanne Amiel, Marlène Rio, Loïc de Pontual et Richard Redon, « Mutations in TCF4, Encoding a Class I Basic Helix-Loop-Helix Transcription Factor, Are Responsible for Pitt-Hopkins Syndrome, a Severe Epileptic Encephalopathy Associated with Autonomic Dysfunction », American Journal of Human Genetics, vol. 80, no 5, , p. 988–993 (ISSN 0002-9297, PMID 17436254, PMCID 1852736, DOI 10.1086/515582, lire en ligne, consulté le )
- ↑ (en) M. Peippo et J. Ignatius, « Pitt-Hopkins Syndrome », Molecular Syndromology, vol. 2, nos 3-5, , p. 171–180 (ISSN 1661-8769, PMID 22670138, PMCID 3366706, DOI 10.1159/000335287, lire en ligne, consulté le )
- 1 2 3 (en) Giuseppe Marangi et Marcella Zollino, « Pitt-Hopkins Syndrome and Differential Diagnosis: A Molecular and Clinical Challenge », Journal of Pediatric Genetics, vol. 4, no 3, , p. 168–176 (ISSN 2146-4596, PMID 27617128, PMCID 4918722, DOI 10.1055/s-0035-1564570, lire en ligne, consulté le )
- 1 2 (en) J. David Sweatt, « Pitt-Hopkins Syndrome: intellectual disability due to loss of TCF4-regulated gene transcription », Experimental & Molecular Medicine, vol. 45, no 5, , e21 (ISSN 2092-6413, PMID 23640545, PMCID 3674405, DOI 10.1038/emm.2013.32, lire en ligne, consulté le )
- ↑ (en) D. Pitt et I. Hopkins, « A syndrome of mental retardation, wide mouth and intermittent overbreathing », Australian Paediatric Journal, vol. 14, no 3, , p. 182–184 (ISSN 0004-993X, PMID 728011, DOI 10.1111/jpc.1978.14.3.182, lire en ligne, consulté le )
Liens externes
- Ressources relatives à la santé :
 Portail de la médecine
Portail de la médecine